Molecular epidemiology


Molecular epidemiology




Back to the
“MOLECULAR PHYLOGENETICS” page
Back to the
“LANE”
introduction page

FACTS
Species:
‣HIV virus
‣Hepatitis delta virus

PUBLICATIONS
‣Journal of Virology, 74: 2525-2532 (2000)
‣Journal of Virology, 78:2537-2544 (2004)
‣Check also ‘Molecular HIV evidence backs accused medics - Expert opinion’ (opinion, not a research paper) in Nature 444: 659-659 (2006)

Related publications from our group on the phylogeny of other groups
‣Check the main “Molecular Phylogenetics” page

PEOPLE INVOLVED
FROM MICHEL
MILINKOVITCH’S LAB
‣Well ... only Michel

MAIN COLLABORATORS
Paul Deny
Institut Fédératif de Recherche Lyon-Est, France.

Investigating a nosocomial infection by HIV-1
Human immunodeficiency virus type 1 (HIV-1) infection risk factors are usually identified directly from a patient’s medical history. However, additional evidence is essential when viral transmission occurs during clinical care or is the object of a criminal trial. Because nucleotide sequences vary greatly among HIV-1 isolates, transmission histories among infected individuals can be investigated through estimated phylogenetic relationships among viral sequences.
Previous studies have shown that phylogenetic methods can lead to accurate reconstruction of known transmission histories and can be used to reconstruct unknown transmission histories. Furthermore, phylogenetic analyses are increasingly used in court and must therefore “rise to a threshold of reliability”.
Investigating (by request of sanitary authorities) a possible health care worker-to-patient transmission of HIV-1 without evidence of blood exposure, we performed extensive phylogenetic analyses. The results presented here unambiguously exclude one HIV-seropositive nurse but strongly suggest another HIV-seropositive nurse as the source of transmission of HIV-1 to the patient.
Our analyses of two distinct genomic regions from free viruses and integrated proviruses combined a large number of geographical and temporal local controls and consistently indicate that HIV-1 sequences from the patient and from nurse 2 share a common ancestor not shared by any other sequence analyzed. This result, strongly supported by a wide range of phylogenetic approaches, indicates that HIV-1 was transmitted from nurse 2 to the patient or vice versa. However, given that nurse 2 had a full Western blot profile and a low CD4+ cell count (indicative of advanced infection) at the time of the patient’s seroconversion, it is very likely that transmission occurred from nurse 2 to the patient. HIV-1 variants with an insertion of five amino acids in the V4 loop quickly emerged in the patient plasma, possibly indicating emergence of mutants escaping immunological neutralization. Insertions in the V4 loop were previously described for a case of mother-to-child transmission. Selection of variants during primary HIV-1 infection is common, and our observation indicates that early samples, preferably from lymphocytes (which usually harbor more ancestral sequences), should be studied when addressing transmission hypotheses.
Although transmission of blood-borne pathogens is a major concern during health care practice, it was suggested that HIV-infected nurses would not be associated with transmission of HIV to a patient because they usually do not perform particularly invasive medical procedures. The case discussed here, indicative of atypical nurse-to-patient HIV-1 transmission, raises the issue of the mandatory screening of nurses. Because such screening would be overwhelmingly cost-ineffective to reduce these rare transmissions, suggestions that are acceptable, from both an ethical and a financial point of view, should be proposed. For example, HIV-infected health care workers, when aware of their seropositivity, could place themselves under the guidance of an expert panel which would determine, on a case-by-case basis, whether practice restrictions are appropriate.
Finally, utmost care is obviously advised in the divulgence of atypical HIV transmissions in a clinical context because it could lead to unnecessary alarm regarding infected medical staff. The case described here may be exceptional, but it indicates nonetheless that safety guidelines established for reducing risks of blood-borne transmission of pathogens between health care workers and patients should be carefully followed.

Reference:
✓Goujon C. P., Schneider V. M., Grofti J., Montigny J., Jeantils V., Astagneau P., Rozenbaum W., Lot F., Frocrain-Herchkovitch C., Delphin N., Le Gal F., Nicolas J.C., Milinkovitch M. C. & P. Dény
Phylogenetic Analyses Indicate an Atypical Nurse-to-Patient Transmission of HIV-1
Journal of Virology, 74: 2525-2532 (2000)
➡ABSTRACT. A human immunodeficiency virus (HIV)-negative patient with no risk factor experienced HIV type 1 (HIV-1) primary infection 4 weeks after being hospitalized for surgery. Among the medical staff, only two night shift nurses were identified as HIV-1 seropositive. No exposure to blood was evidenced. To test the hypothesis of a possible nurse-to-patient transmission, phylogenetic analyses were conducted using two HIV-1 genomic regions (pol reverse transcriptase [RT] and env C2C4), each compared with reference strains and large local control sets (57 RT and 41 C2C4 local controls). Extensive analyses using multiple methodologies allowed us to test the robustness of phylogeny inference and to assess transmission hypotheses. Results allow us to unambiguously exclude one HIV-positive nurse and strongly suggest the other HIV-positive nurse as the source of infection of the patient.
➡REMARK. Utmost care is obviously advised in the divulgence of atypical HIV transmissions in a clinical context because it could lead to unnecessary alarm regarding infected medical staff.
➡Check also the opinions of multiple epidemiologists and phylogeneticists backing up a Nature paper published in 2006 (‘Molecular Epidemiology: HIV-1 and HCV sequences from Libyan outbreak’ by de Oliveira et al., Nature 444, 836-837): this study provided a firm alibi for the six medical workers who once faced the death penalty in Libya (the Bulgarian and Palestinian workers had been charged with deliberately infecting more than 400 children with HIV in 1998). De Oliveira used the genetic sequences of the viruses isolated from the patients to reconstruct the exact phylogeny of the outbreak. They showed that the strain of HIV with which the children had been infected was already present and spreading locally in the mid-1990s, long before the medics arrived in Libya in 1998. They were freed in July 2007 after 8 years in a Libyan prison.
✓Lemey P., Hillis D.M., Gonzalez Candelas F., Milinkovitch M.C., and T. Leitner
Molecular HIV evidence backs accused medics - Expert opinion
Nature (‘opinion’, not a research paper) 444: 659-659 (2006)
Uncovering an ancient African radiation of hepatitis D virus
Hepatitis D virus (HDV) is a transmissible agent discovered in 1977 that requires helper functions from the hepatitis B virus (HBV) for virion assembly and propagation. Thus, HDV infection is necessarily associated with HBV infection because HDV ribonucleoprotein buds through the hepatitis B surface antigen (HBsAg) excretory pathway. The HDV genome is a circular single-stranded RNA genome of approximately 1,680 bases with extensive intramolecular complementarity. Part of the HDV genome might have historical homology to viroids or plant virus satellite RNA sequences, and a rolling-circle model has been developed for viral RNA replication. HDV mRNA is translated to sHD and LHD proteins, corresponding respectively to the “small-p24” and the “large-p27” hepatitis Delta proteins. sHD is required for viral replication and might promote RNA polymerase II elongation of nascent HDV RNA, while LHD is essential for HDV particle assembly.
HDV-HBV coinfection and HDV superinfection of a patient chronically infected by HBV both lead to a liver disease more severe than that induced by HBV alone. HDV is highly endemic in Mediterranean countries, the Middle East, Central Africa, and northern parts of South America. In contrast, in industrialized countries, its prevalence is low and its transmission is often associated with intravenous drug use. Intrafamilial transmission has been described in southern Italy, which has been considered an area of high endemicity.
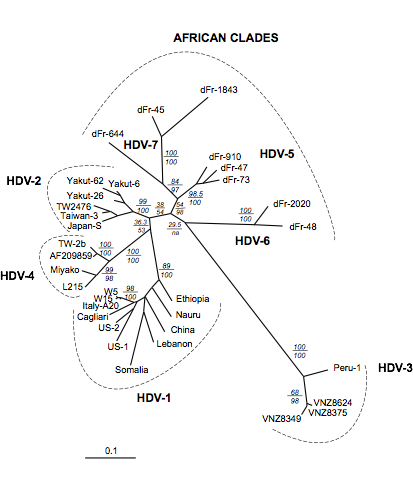
Figure: ML analyses of 33 sHD cDNA sequences. Analyses were conducted using PAUP4b6 and MetaPIGA. Numbers above the horizontal lines indicate bootstrap values (100 replicates with PAUP*, parameters constrained to those obtained from the ML analysis of the original data set), and those below the lines indicate metaGA posterior probabilities (10,000 samples). Scale is in percent expected substitution per position. Suggested clade names are indicated.
Reference:
✓Radjef N., Gordien E., Ivaniushina V., Gault E., Anaïs P., Drugan T., Trinchet J.C., Roulot D., Tamby M., Milinkovitch M.C. & P. Dény
Molecular Phylogenetic Analyses Indicate a Wide and Ancient Radiation of African Hepatitis Delta Virus, Suggesting a Deltavirus Genus of at Least Seven Major Clades
Journal of Virology, 78:2537-2544 (2004)
➡ABSTRACT. Hepatitis D virus (HDV) is a satellite of hepatitis B virus (HBV) for transmission and propagation and infects nearly 20 million people worldwide. The HDV genome is a compact circular single-stranded RNA genome with extensive intramolecular complementarity. Despite its different epidemiological and pathological patterns, the variability and geographical distribution of HDV are limited to three genotypes and two subtypes that have been characterized to date. Phylogenetic reconstructions based on the delta antigen gene and full-length genome sequence data show an extensive and probably ancient radiation of African lineages, suggesting that the genetic variability of HDV is much more complex than was previously thought, with evidence of additional clades. These results relate the geographic distribution of HDV more closely to the genetic variability of its helper HBV.
➡REMARK. This wide African radiation of HDV was confirmed and further characterized in the following years.
